
When not to use machine learning a perspective on potential and limitations Matthew R. Carbone Computational Science Initiative Brookhaven National Laboratory Upton New York 11973
2025-04-29
0
0
329.05KB
7 页
10玖币
侵权投诉

When not to use machine learning: a perspective on potential and limitations
Matthew R. Carbone∗
Computational Science Initiative, Brookhaven National Laboratory, Upton, New York 11973
(Dated: October 7, 2022)
The unparalleled success of artificial intelligence (AI) in the technology sector has catalyzed an
enormous amount of research in the scientific community. It has proven to be a powerful tool, but
as with any rapidly developing field, the deluge of information can be overwhelming, confusing and
sometimes misleading. This can make it easy to become lost in the same hype cycles that have
historically ended in the periods of scarce funding and depleted expectations known as AI Winters.
Furthermore, while the importance of innovative, high-risk research cannot be overstated, it is also
imperative to understand the fundamental limits of available techniques, especially in young fields
where the rules appear to be constantly rewritten and as the likelihood of application to high-stakes
scenarios increases. In this perspective, we highlight the guiding principles of data-driven modeling,
how these principles imbue models with almost magical predictive power, and how they also impose
limitations on the scope of problems they can address. Particularly, understanding when not to use
data-driven techniques, such as machine learning, is not something commonly explored, but is just as
important as knowing how to apply the techniques properly. We hope that the discussion to follow
provides researchers throughout the sciences with a better understanding of when said techniques
are appropriate, the pitfalls to watch for, and most importantly, the confidence to leverage the power
they can provide.
Keywords: artificial intelligence; computation/computing; machine learning; modeling
I. INTRODUCTION
The story of artificial intelligence (AI) began in the
mid 1900’s, when computer scientists started considering
a simple question: “can machines think?” [1,2]. Ever
since, the mythical goal of achieving true “human-like”
AI has framed decades of scientific conversation and has
motivated many key breakthroughs, including the back-
bone of modern machine learning (ML) algorithms: neu-
ral networks [3–5]. As the computational machinery re-
quired to realize the practical applications of ML would
come decades later [6], its full potential would not yet
be immediately understood. That time has come, and
despite the turbulence of multiple “AI winters” over the
past decades [7–9], we are currently living the AI/ML
revolution.
Broadly, AI and ML are two related families of meth-
ods that fall under the larger “data-driven” umbrella.
Built upon well established theory in the statistics and
applied mathematics communities [4], modern-day AI
and ML is best understood as the intersection of powerful
modeling paradigms with “big-data” and bleeding edge
hardware (e.g. graphics processing units; GPUs). The
general interpretation (though not the only one [10]) is
that AI is a superset of ML [11] and consists of techniques
that are used to mimic human cognition and decision-
making, whereas ML is more focused on the mathemati-
cal and numerical approaches. Often, ML is described as
the ability of a program to learn a task without being pro-
grammed with task-specific heuristics [12]. However, the
distinction between AI and ML is not germane to most
∗mcarbone@bnl.gov
applications (and many applications use parts of both),
hence the blanket term “AI/ML” is used commonly as
a substitute for “data-driven” in many contexts. In this
perspective, we will focus primarily on supervised ML,
though many of the key points to come apply to data-
driven approaches in general.
Cruising behind the slipstream created by tremendous
success in the technology sector, ML has found wide ap-
plicability in the materials, chemical and physical sci-
ences. For example, the discovery, characterization and
design of new materials, molecules and nanoparticles [13–
24], surrogate models for spectroscopy and other proper-
ties [25–28], self-driving laboratories/autonomous experi-
mentation [29–34], and neural network potentials [35–39]
have all been powered by ML and related methods. The
current state of ML in materials science specifically has
also been thoroughly documented in many excellent re-
views [15,40–42] that cover subject matter ranging from
applications to computational screening and interpreta-
tion. On a related note, for technical details and timely
tutorials, we refer those interested readers to Refs. 43
and 44. However, while the scope of ML-relevant prob-
lems is huge, not every problem can effectively leverage
the power ML provides. Worse still, sometimes ML may
seem to be a perfectly reasonable choice only to fail dra-
matically [45]; such failures can often be traced back to
the foundations of any ML tool: the data.
In this perspective, we ask and answer a foundational
question which ultimately has everything to do with
data: when should you not use machine learning? ML
is the jackhammer of the applied math world, and is
able to channel incredible power provided by the inter-
play of highly flexible models, large databases and GPU-
enabled supercomputers. But you wouldn’t use a jack-
hammer to do brain surgery. At least for the time being,
arXiv:2210.02666v1 [cs.LG] 6 Oct 2022
2
there are classes of problems for which ML is not well-
suited [43,46]. We address this issue not to dissuade
researchers from using these methods, but rather to em-
power them to do so correctly, and to avoid wasting valu-
able time and resources. Understanding the limitations
and application spaces of our tools will help us build bet-
ter ones, and solve larger problems more confidently and
with more consistency.
II. THE DEVIL’S IN THE DISTANCE
Newcomers to the field of ML will find themselves im-
mediately buried under an avalanche of enticing algo-
rithms applicable to their scientific problem [47]. Many of
these choices are so sophisticated that it is unreasonable
to expect any ML non-expert to understand their finer
nuances and how/why they can fail. The steep learning
curve combined with their intrinsic complexity, mythical
“black box” nature and stunning ability to make accurate
predictions can make ML appear almost magical. It may
come as a surprise then that in spite of said complexi-
ties, almost all supervised ML models are paradigmat-
ically the same and are built upon a familiar quantity:
distance.
The supervised ML problem is one of minimizing the
distance between predicted and true values mapped by
an approximate function on the appropriate inputs. A
distance can be a proper metric, such as the Euclidean
or Manhattan norms, or something less pedestrian, such
as a divergence (a distance measure between two dis-
tributions) or a cross-entropy loss function. Regardless,
the principle is the same: consider un-regularized, super-
vised ML, where given a source of ground truth Fand
a ML model fθ,the goal is to find parameters θsuch
that the distance between F(x) and fθ(x) is as small as
possible for all possible xin some use case. While this
is only one type of ML, most techniques share this com-
mon theme. For example, Deep Q reinforcement learn-
ing [48] leverages neural networks to map states (inputs)
to decisions (outputs), and unsupervised learning algo-
rithms rely on the same notion of distance to perform
clustering and dimensionality reduction that supervised
learning techniques use to minimize loss functions. Vari-
ational autoencoders [49–51] try not only to minimize re-
construction loss, but simultaneously keep a compressed,
latent representation as close to some target distribution
as possible (usually for use in generative applications).
Numerical optimization is the engine that systematically
tunes model parameters θin gradient-based ML,1and its
only objective is to minimize some measure of distance
between ground truth and model predictions.
1The classic numerical optimizer is gradient/stochastic gradi-
ent descent, with more recently established developments showing
systematic improvements in deep learning, e.g. Adam [52].
Additionally, in order to increase the confidence that
ML models will be successful for a given task, it helps
if the desired function is smooth: i.e. a small change in
a feature should ideally correspond to a relatively small
change in the target. This idea is more readily defined
for regression than for classification, and the data be-
ing amenable to gradient-based methods is is not strictly
required for ML to be successful. For example, e.g. Ran-
dom Forests are not generally trained using gradient-
based optimizers, but satisfying this requirement will
usually help models generalize more effectively. The dis-
tance between the features of any two points of data is
informed entirely by their numerical vector representa-
tion, and while these representations can be intuitive or
human-interpretable, they must be mathematically rig-
orous.
The devil here, so to speak, is that what might appear
intuitive to the experimenter may not be to the machine.
For example, consider the problem of discovering novel
molecules with certain properties. Molecules can be first
encoded in string format (e.g. SMILES [53]), and then
a numerical latent representation. The structure of this
latent space is informed by some target property [16], and
because any point in the latent space is just a numeric
vector living in a vector space, a distance can be easily
defined. This powerful encoding method can be used to
“interpolate between molecules” and thus discover new
ones that perhaps we haven’t previously considered, but
it still relies on the principle of distance, both between
molecules in the compressed latent space, and their target
properties.
Concretely, the length scales for differentiating be-
tween data points in the feature space are set by the
corresponding targets. Large changes in target values
between data points can cause ML models to “focus” on
the changes in the input space that caused it, possibly
at the expense of failing to capture small changes. This
is often referred to as the bias-variance trade-off. Most
readers may be familiar with the concept of over-fitting:
for instance, essentially any set of observations can be
fit exactly by an arbitrarily high-order polynomial, but
doing so will produce wildly varying results during infer-
ence and be unlikely to have captured anything meaning-
ful about the underlying function. Conversely, a linear fit
will only capture the most simple trends to the point of
being useless for any nonlinear phenomena. Fig. 1show-
cases a common middle ground, where the primary trend
of the data is captured by a Gaussian Process [54], and
smaller fluctuations are understood as noisy variations
around that trend.
Consider a more realistic example: the Materials
Project [55] database contains many geometry-relaxed
structures, each with different compositions, space
groups and local symmetries at 0 Kelvin. Thus, within
this database, changes in e.g. the optical properties of
these materials is primarily due to the aforementioned
structural differences and not due to thermal disorder
(i.e. distortions) one would find when running a molec-

摘要:
展开>>
收起<<
Whennottousemachinelearning:aperspectiveonpotentialandlimitationsMatthewR.Carbone*ComputationalScienceInitiative,BrookhavenNationalLaboratory,Upton,NewYork11973(Dated:October7,2022)Theunparalleledsuccessofarti cialintelligence(AI)inthetechnologysectorhascatalyzedanenormousamountofresearchinthescient...
声明:本站为文档C2C交易模式,即用户上传的文档直接被用户下载,本站只是中间服务平台,本站所有文档下载所得的收益归上传人(含作者)所有。玖贝云文库仅提供信息存储空间,仅对用户上传内容的表现方式做保护处理,对上载内容本身不做任何修改或编辑。若文档所含内容侵犯了您的版权或隐私,请立即通知玖贝云文库,我们立即给予删除!
相关推荐
-
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包05专题五 走进社会生活 遵守社会规则VIP免费

 2024-11-21 24
2024-11-21 24 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包05专题五 走进社会生活 遵守社会规则VIP免费

 2024-11-21 22
2024-11-21 22 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包03专题三 青春时光 做情绪情感的主人VIP免费
 2024-11-21 16
2024-11-21 16 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包03专题三 青春时光 做情绪情感的主人VIP免费

 2024-11-21 19
2024-11-21 19 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包02专题二 友谊的天空 师长情谊VIP免费
 2024-11-21 19
2024-11-21 19 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包02专题二 友谊的天空 师长情谊VIP免费
 2024-11-21 20
2024-11-21 20 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包01专题一 成长的节拍 生命的思考VIP免费
 2024-11-21 23
2024-11-21 23 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包01专题一 成长的节拍 生命的思考VIP免费
 2024-11-21 24
2024-11-21 24 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包《53中考》全国道德与法治资料包VIP免费
 2024-11-21 27
2024-11-21 27 -
曲一线系列初中《5中考3年模拟》2023专题解释全国道德与法治资料包07专题七 坚持宪法至上 崇尚法治精神VIP免费
 2024-11-21 18
2024-11-21 18
分类:图书资源
价格:10玖币
属性:7 页
大小:329.05KB
格式:PDF
时间:2025-04-29
作者详情

相关内容
-

2025届重庆市西南大学附属中学高三下学期5月全镇模拟物理试题(含答案)
分类:中学教育
时间:2025-12-31
标签:无
格式:PDF
价格:10 玖币
-

2025届重庆市西南大学附属中学高三下学期5月全镇模拟化学试题(含答案)
分类:中学教育
时间:2025-12-31
标签:无
格式:PDF
价格:10 玖币
-
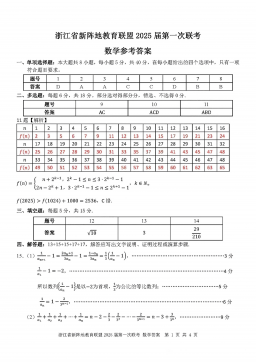
2025届浙江省新阵地联盟高三10月联考数学答案
分类:中学教育
时间:2025-12-31
标签:无
格式:PDF
价格:10 玖币
-

2025届重庆市西南大学附属中学高三下学期5月全镇模拟数学试题(含答案)
分类:中学教育
时间:2025-12-31
标签:无
格式:PDF
价格:10 玖币
-

2025届重庆康德三诊英语+答案
分类:中学教育
时间:2026-01-03
标签:无
格式:PDF
价格:10 玖币

